New Drugs (Originator drugs) are medical drugs which are clearly differentiated in active ingredients, dosage, administration, indication and efficacy from drugs which have already been approved. New Drugs mainly consist of drugs containing new active ingredients, new ethical combination drugs, drugs with a new administration route, drugs with a new indication, drugs in new dosage forms, and new dosage drugs.
6.1 Pharmaceuticals
Definition of New Drugs
Pharmaceutical Product Approvals
To manufacture, distribute and sell new drugs, pharmaceutical companies must first obtain marketing approval from the PMDA. Approvals are made by the PMDA based on an evaluation of the new drug’s efficacy and safety information obtained via clinical trials. Clinical trial information and filing documents must conform to the quality, GLP and GCP requirements defined by the MHLW.
In addition to the standard approval process, the MHLW has implemented to mechanisms for expedited reviews and approvals of innovative pharmaceutical products.
To address the development lag between Japan and other major markets, the MHLW implemented the “Sakigake” review policy in 2014 that awards prioritized reviews and expedited approvals for products that are developed for and filed for first in Japan. To be eligible for “Sakigake” reviews, new drugs must meet all of the following criteria: 1) innovative new mechanism of action, 2) degree of severity of the target indication, 3) high degree of effectiveness, and 4) a desire to develop and file first in Japan.
Furthermore, In 2017, the MHLW implemented the “Conditional Expedited Approval” Policy for drugs that address disease states with small patient populations with limited treatment options and large unmet medical needs for which the execution of confirmatory clinical trials are difficult. The policy enables pharmaceutical companies to file for approval with efficacy and safety data generated from non-confirmatory trials, such as phase II trials, on the condition that additional safety and surveillance measures, including the use of real world data, are implemented. It is expected that this policy will support the development of novel treatments including iPS cell based genetic therapies.
Pharmaceutical Reimbursement Pricing
Once marketing approval is obtained, manufacturers must apply for reimbursement pricing under the National Health Insurance (NHI) system. The NHI reimbursement price is the price at which payors reimburse medical institutions for pharmaceutical products used to treat patients.
Initial NHI Reimbursement prices that are set by the MHLW’s Central Medical Council based primarily on the comparable based method when similar approved products already exist on the market, or the cost based method for novel therapeutics with no similar approved products.
In addition, the MHLW has also implemented pricing premiums to both incentivize the development of innovative products while constraining the reimbursement prices of products whose degree innovativeness are assessed to be limited. Products with superior efficacy or usefulness compared to existing products, with orphan or pediatric indications, or for which Japan is the first country are eligible for additional price premiums, whether priced via the comparator or the cost based methods
Once set, pharmaceutical reimbursement prices are revised biannually based on an assessment of the variance between NHI reimbursement prices and actual market (wholesale) prices. While the price difference between NHI reimbursement prices and actual market prices enable medical institutions and pharmacies to book revenue, biannual price revisions are intended to limit discounting to within a reasonable range known as the “R-zone” established by the MHLW, currently set at 2% of reimbursement prices. In practice, the biannual price revisions have the effect of lowering drug prices, enabling the MHLW to control the natural growth in overall healthcare spending.
In addition to the standard biennial reassessment of drug prices in Japan, additional adjustments have also been implement to ensure products are profitable and to promote the development of new drugs while limiting overall costs by reducing prices for drugs that have seen significant market expansion or that have lost patent protection.
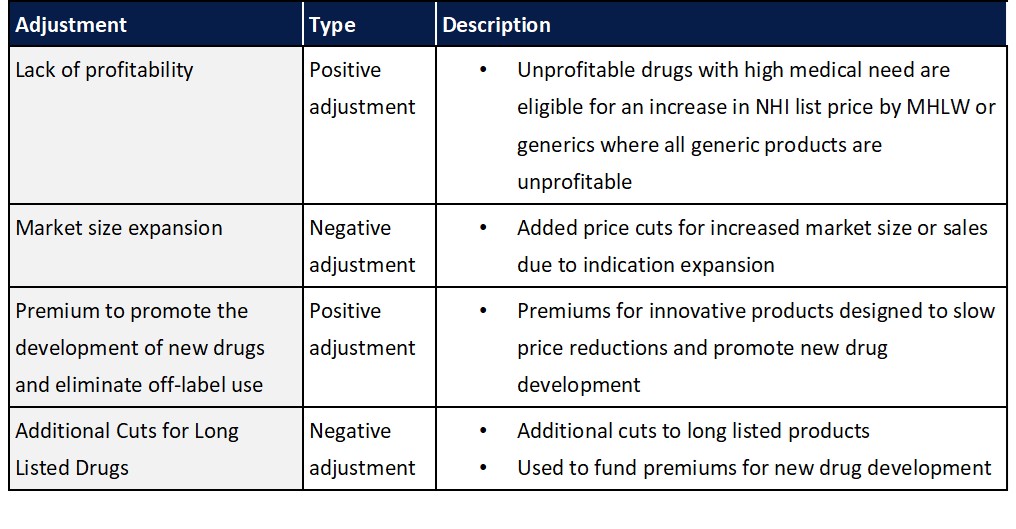
As healthcare costs continue to rise, it is expected that additional revisions to pharmaceutical reimbursement pricing mechanism may be made going forward. While undetermined, to date, topics such as the introduction of health economics outcome considerations when setting reimbursement prices in addition to potential shift towards annual price revisions have been discussed.
Pharmaceutical Product Quality and Safety
Similar to other major markets, the development, manufacture and distribution of pharmaceutical products in Japan are also governed by GXP regulations such as GLP, GCP, GMP and GVP standards. However, there are a few notable differences in Japanese regulations and policies related to product quality and safety.
First, under the Pharmaceutical Affairs Law, to comply with Japan’s Good Quality Practices (GQP) and GVP requirements, pharmaceutical companies must designate three individuals to take on the following roles unique to Japan to oversee and govern product quality and safety related activities:
- General marketing compliance officer – Individual with overall responsibility for product quality and safety
- Quality assurance manager – Individual responsible to ensure quality activities comply with GQP regulations
- Safety manager – Individual responsible for ensure safety activites comply with GVP regulations
Second, upon approval of novel drugs, Japan requires pharmaceutical companies to conduct post marketing surveillance activities to track and ensure patient safety. Surveillance activities are generally required to be conducted during the 6 month period after launch and are intended to identify track any adverse events in the real world setting that may not have been observed during clinical trials. These activities are governed by Japan’s Good Post-marketing Surveillance Practice (GPSP) regulations.
Third, to further ensure patient safety, new pharmaceutical products are generally restricted to 14 day prescriptions for one year from the first day of the month pricing is listed. Exceptions to the 14 day prescription restriction for specific new drugs can be awarded by the Central Medical Council for products that meet either of the following two criteria:
- New drugs (e.g. compound drugs) with APIs that have the same effect, efficacy, dosing and administration as approved products with over one year of clinical experience
- New drugs where, based on its characteristics or the disease state, only over 14 day formulations exist and the safety of over 14 day administration has been confirmed