6.1新医薬品
新医薬品とは
医療用医薬品の承認
製薬会社が新医薬品を製造、配送、販売するためには、まずPMDA(独立行政法人医薬品医療機器総合機構)から販売承認を得る必要がある。PMDAは、臨床試験を通じて得られた新医薬品の効果と安全性への評価情報に基づいて承認をする。臨床試験データ及び提出される書類の書式は、厚生労働省(以下、厚労省)が定める品質、優良試験所基準(GLP: Good Laboratory Practice) 、薬品優良臨床試験基準(GCP: Good Clinical Practice)に従ったものでなければならない。[1]
厚労省は、通常の承認プロセスに加えて、画期的な新薬の開発を促進できるよう、優先的に審査・承認する制度も導入している。
患者に世界で最先端の治療薬を最も早く提供することを目指し、厚労省は2014年、日本で開発され、日本で初めて登録申請される製品への審査と承認を優先的かつ短期間で行う「先駆け審査指定制度」を導入した。「先駆け審査指定制度」の対象となるためには、新医薬品は次の条件を満たさなければならない:1)革新的で新しい作用機序、2)対象とする適応症の重篤度、3)極めて高い効率性、4)世界に先駆けて日本で開発し始めて登録申請する意図がある。[2]
さらに2017年厚労省は、患者数が少なく治療の選択肢が限られ、検証臨床試験の実施が難しいために大きな医療上の満たされないニーズがある疾病への治療薬を対象とする「医薬品条件付早期承認制度」を導入した。この制度により製薬企業は、製販後に有効性・安全性の再確認等のために必要な調査等を実施すること等を承認条件として、フェーズ2治験[3]などの検証的臨床試験以外の臨床試験等、得られた一定程度の有効性及び安全性に関するデータを基に承認申請を行えるようになった。本制度は、iPS細胞を利用した遺伝子治療を含む新たな治療法の開発を支援すると期待されている。[4]
医療用医薬品の薬価
製造者は販売承認を得たら、次に薬価基準収載希望書を厚生労働大臣宛に提出し承認を得る。その後に薬価基準へ収載され、保険適用となる。日本の医療保険制度においては、患者の治療に使われた医薬品の対価の一部を医療機関に払い戻す。その払い戻し額を薬価と呼ぶ。
収載当初における薬価は、厚労省の中央社会保険医療協議会が、市場に既に承認された類似薬が存在する場合は比較方式で、承認された類似薬が存在しない新薬の場合は、原価計算方式で決定する。[5]
加えて厚労省は、革新性が限定的と評価された医薬品の薬価を抑制しつつ革新性の高い医薬品開発を奨励する両方の目的で、薬価への補正加算制度も導入している。既存品に比べて優れた有効性や有用性を持つ、希少疾病や小児への適応、または日本で初めて登録申請される医薬品については、比較方式と原価計算方式のいずれの場合であっても補正加算の対象となる。
薬価収載された後、医療用医薬品の薬価は実際の市場価格とのかい離率を基に、1年に1度、定期的に改定される。薬価と実勢価格のかい離は医療機関や薬局の収益となっており、厚労省は流通安定のための最小限必要な調整比率を許容される割引率を「R幅」と定め、薬価の2%と設定し、かい離率がこの2%を超えた場合、薬価が改訂される仕組みを導入している。実際、薬価改定には医薬品の価格を引き下げる効果があり、それによって厚労省は全体的な医療費支出が自然増となることを防いでいる。
日本においては、このような標準的な薬価改定に加えて、市場が大幅に拡大したり特許切れとなったりした医薬品の価格を下げて医療費全体の増加を抑制する一方、医薬品の利益率を確保して新薬の開発を奨励するために、追加の再算定を行う制度も定めている。
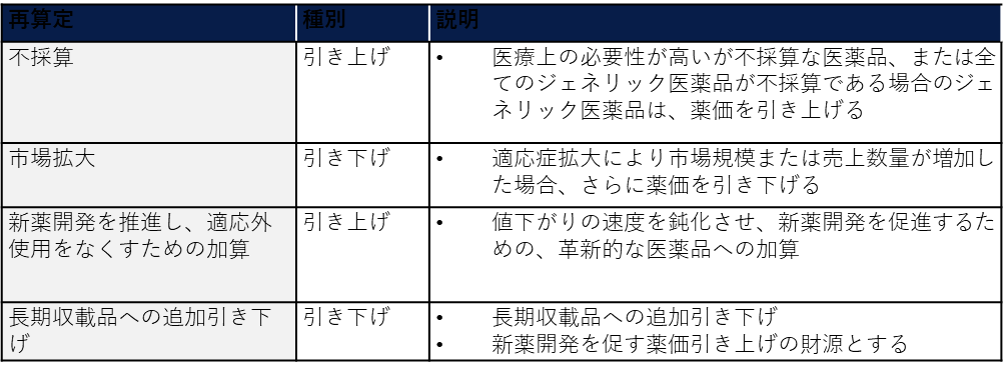
医療費が増大を続ける中で、今後さらなる薬価決定制度の変更が行われることが予測される。現時点では、薬価決定において医療経済学的視点を考慮する制度の導入などの論点が協議されている。
医療用医薬品の安全性と品質
他の主要市場と同様に、日本における医薬品の開発、製造、販売は、GLP、GCP、製造所における製造管理、品質管理の基準(GMP:Good Manufacturing Practice)、製造販売後安全管理の基準(GVP:Good Vigilance Practice)等を含む医薬品の開発から工場管理等までの薬事規制に関する各基準(GXP: Good x practice)によって統制されている。しかし、製品の品質及び安全性に関して、日本の法律及び政策には、他国と比較していくつかの顕著な違いがある。
初めに、薬機法の下で日本の医薬品・医薬部外品、化粧品及び再生医療など製品の品質管理の基準(GQP:Good Quality Practice)とGVP要件を満たすために、製薬会社は製品の品質と安全性にかかわる活動を監督し統制するために、日本に特有の以下の役割を果たすために3名の責任者を任命しなければならない。[6]
- General marketing compliance officer (総括製造販売責任者):製品の品質及び安全性について全体責任を負う
- Quality assurance manager (品質保証責任者):GQPの規則に従って、品質関連の活動が行われることを確実にする責任を負う
- Safety manager (安全管理責任者):GVPの規則に従って、安全関連の活動が行われることを確実にする責任を負う
2つ目に、新薬が承認された後、厚労省は製薬会社に、患者の安全性を追跡し確保するために医薬品の製造販売後の調査活動を行うことを義務付けている。調査活動は通常、新薬の上市後6ヵ月間実施することが義務付けられており、臨床試験中には観察されなかった可能性のある何らかの有害事象が現実の使用において起きていないかどうかを追跡することを目的としている。これらの業務は、日本の医薬品製造販売後調査・試験の実施の基準(GPSP:Good Post-marketing Surveillance Practice )に準拠して行われている。[7]
3つ目に、さらに患者の安全を確実にするため、新薬は一般的に、収載された月の初日から1年間は処方日数が14日までに制限される。だが、以下2つの基準のいずれかを満たす医療用医薬品の場合は、14日の処方日数上限を適用しない特例が、中央医療協議会によって認められている。[8]
- 1年以上の臨床使用経験がある既収載品と効能・効果、用量・用法の有効成分を配合した新薬(例えば新医療用配合剤等)
- 疾病の特性や製剤上の特性により、含有量が14日を超える製剤のみが存在しており、投薬期間が14日を超えることの安全性が確認されている新薬
参考文献
[1] 医薬基盤研究所「医薬品・バイオ研究の実用化に向けて~知っておきたい薬事規制~」https://www.nibiohn.go.jp/guide/page1.html(アクセス2018年2月1日)
[2] 厚生労働省「先駆け審査指定制度について」http://www.mhlw.go.jp/seisakunitsuite/bunya/kenkou_iryou/iyakuhin/topics/tp150514-01.html (アクセス日2018年2月1日)
[3] 国立がん研究センター「研究段階の医療(臨床試験、治験など)詳細情報」https://ganjoho.jp/med_pro/med_info/ct/ct_details.html (アクセス日2018年10月4日)
[4] 厚生労働省「医薬品の条件付き早期承認制度の実施について」https://www.pmda.go.jp/files/000220723.pdf(アクセス日2018年2月1日)
[5] 厚生労働省「日本の薬価制度について」http://www.mhlw.go.jp/file/04-Houdouhappyou-11123000-Iyakushokuhinkyoku-Shinsakanrika/0000135596.pdf(アクセス2018年2月1日)
[6] 厚生労働省「医薬品の製造販売業者における三役の適切な業務実施に関する留意事項(案)」http://www.mhlw.go.jp/file/05-Shingikai-10601000-Daijinkanboukouseikagakuka-Kouseikagakuka/iyakuhinniryoukikiseidobukai2sannkou6-2.pdf(アクセス日 2018年1月20日)
[7] PMDA「市販直後調査に関する情報」http://www.pmda.go.jp/review-services/drug-reviews/review-information/p-drugs/0006.html(アクセス日 2018年1月20日)
[8] 厚生労働省「新医薬品の14日間処方日数制限の見直しについて」http://www8.cao.go.jp/kisei-kaikaku/suishin/meeting/wg/iryou/20171106/171106iryou03.pdf(アクセス日 2018年2月1日)